The specific heat capacity of a pure substance or a mixture is conventionally defined as the heat required to raise the temperature of 1 mole of the substance under specified conditions. However, formally, it is defined as the limit of the ratio d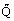 /dT as dT, the temperature change, and d
/dT as dT, the temperature change, and d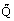 , the heat input, go to zero. It is possible to define several heat capacities depending upon the conditions and we consider first the heat capacity at constant volume
, the heat input, go to zero. It is possible to define several heat capacities depending upon the conditions and we consider first the heat capacity at constant volume  . In this circumstance, the heat is supplied to the substance, presumed a fluid, in a rigid container of fixed volume. The thermodynamic system is a closed one and the appropriate form of the first law of thermodynamics is
. In this circumstance, the heat is supplied to the substance, presumed a fluid, in a rigid container of fixed volume. The thermodynamic system is a closed one and the appropriate form of the first law of thermodynamics is

where dũ is the change of internal energy and  the work done. But since
the work done. But since 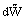 = pext
= pext and
and 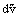 is zero, the work done is zero and
is zero, the work done is zero and

where  is the entropy.
is the entropy.
The realization of a second specific, molar heat capacity, that at constant pressure, for a liquid or a solid, can be performed merely by supplying heat to the sample under a constant pressure, p. The appropriate first law is still the same but

is not zero so

but because  we can readily show that
we can readily show that
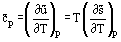
It is also possible to define a specific molar heat capacity at saturation,  , which is related to the amount of heat that must be supplied to 1 mole of the system for a unit temperature change while maintaining the phase equilibrium along the saturation line. The heat capacity and saturation can be written
, which is related to the amount of heat that must be supplied to 1 mole of the system for a unit temperature change while maintaining the phase equilibrium along the saturation line. The heat capacity and saturation can be written
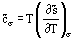
The three heat capacities may be interrelated by means of thermodynamic relationships involving the equation of state of the material [Bett et al. (1975)]. The three heat capacities are functions of the thermodynamic state and it is usual to express  as a function of p and T, and
as a function of p and T, and 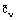 as a function of
as a function of  and T.
and T.
Measurements of the heat capacity of solids and liquids are rather easily performed routinely with a very modest accuracy (a few percent) with modern differential scanning calorimeters [Grolier (1994)]. However, since for many engineering purposes, one is interested in enthalpy differences between streams with enthalpies of a similar magnitude, such an accuracy is often not adequate. Considerable effort has been devoted to the development of instruments for the measurement of heat capacities. Such instruments are almost always electrically operated calorimeters. In almost all calorimeters a known amount of substance is heated electrically and the temperature rise measured. The system in which this is done can be either closed (for solid, liquid or gas) or steady-state flow (liquid or gas). A comprehensive review of classical methods has been given by McCullough and Scott (1968). Techniques for the measurement of the heat capacity of liquids have recently been reviewed by Grolier (1994) in the context of a wider review of calorimetry.
The isobaric heat capacity and the isochoric heat capacity are related by the equation
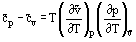
which, for an ideal gas, leads to the simple result

For the ideal gas state, the heat capacity may be expressed through statistical mechanics in terms of the contributions to the translational and internal energies of the molecules [Rushbrooke (1949)]. In turn, some of the internal contribution arising from rotational, vibrational and electronic modes of motion can often then be determined from spectroscopic measurement of the frequencies of the normal mode of motion of the molecule. For many molecules, this process provides a more accurate means of determining the ideal-gas heat capacity of the material than does direct measurement [de Reuck and Craven (1993)].
As the density is increased from the ideal gas state, the energy of the ensemble of molecules acquires a component arising from the interactions between molecules (the configurational part) and this cannot be evaluated theoretically for any but the simplest molecules so that the only source of information on the heat capacity is then from direct or indirect measurement. When there are no measurements available it is necessary to have recourse to estimation methods [Reid et al. (1975)].
REFERENCES
Bett, K. E., Rowlinson, J. S., and Saville, G. (1975) Thermodynamics for Chemical Engineers, Athlone, London. DOI: 10.1016/0300-9467(76)80049-4
de Reuck, K. M. and Craven, R. J. B. (1993) International Thermodynamic Tables of the Fluid State—12: Methanol, Blackwell Scientific, Oxford.
Grolier, J.-P. (1994) Heat Capacity of Organic Liquids in Experimental Thermodynamics IV, Solution Calorimetry, K. N. Marsh and P. A. G. O'Hare, eds., Blackwell Scientific, Oxford.
McCullough, J. P. and Scott, D. W. (1968) Experimental Thermodynamics 1, Calorimetry of Non-Reacting Systems, Butterworths, London.
Reid, R. C, Prausnitz, J. M., and Sherwood, T. K. (1977) The Properties of Gases and Liquids, McGraw-Hill, New York.
Rushbrooke, G. S. (1949) Introduction to Statistical Mechanics, Clarendon, Oxford.
Referencias
- Bett, K. E., Rowlinson, J. S., and Saville, G. (1975) Thermodynamics for Chemical Engineers, Athlone, London. DOI: 10.1016/0300-9467(76)80049-4
- de Reuck, K. M. and Craven, R. J. B. (1993) International Thermodynamic Tables of the Fluid State—12: Methanol, Blackwell Scientific, Oxford.
- Grolier, J.-P. (1994) Heat Capacity of Organic Liquids in Experimental Thermodynamics IV, Solution Calorimetry, K. N. Marsh and P. A. G. O'Hare, eds., Blackwell Scientific, Oxford.
- McCullough, J. P. and Scott, D. W. (1968) Experimental Thermodynamics 1, Calorimetry of Non-Reacting Systems, Butterworths, London.
- Reid, R. C, Prausnitz, J. M., and Sherwood, T. K. (1977) The Properties of Gases and Liquids, McGraw-Hill, New York.
- Rushbrooke, G. S. (1949) Introduction to Statistical Mechanics, Clarendon, Oxford.